







Sickle cell retinopathy (SCR) is a progressive, sight-threatening ocular complication of sickle cell disease (SCD).1 Advances in systemic care have significantly increased the longevity of individuals with SCD, resulting in a growing population at risk for the disease’s chronic, end-organ complications, including retinopathy.2 This shift underscores the need for retina specialists to have a thorough understanding of SCR, from its distinct pathophysiology to both established and emerging therapeutic approaches. This article reviews current knowledge of SCR, with an emphasis on risk stratification, classification, and the evolving management landscape.
Epidemiology and Genotype-Based Risk
Sickle cell disease, an autosomal recessive disorder stemming from mutations in the HBB gene, affects more than 100,000 people in the United States and an estimated 7.74 million globally.2 Although the disease is most prevalent in individuals of African, Mediterranean, or Indian descent, its clinical expression and associated ocular risk are highly dependent on genotype.3 The most common genotypes are homozygous hemoglobin SS (HbSS), or sickle cell anemia, and the compound heterozygous state hemoglobin SC (HbSC).4
Although HbSC is generally considered the milder genotype systemically, it carries a significantly higher risk of developing more severe proliferative sickle cell retinopathy (PSCR) than HbSS.5 This counterintuitive trend, termed the “sickle cell paradox,” is exemplified in a 20-year prospective Jamaican cohort study, which found that 43% of HbSC patients developed PSCR by their mid-20s (ages 24 to 26) vs 14% of HbSS patients.6 This disparity is attributed to differences in blood rheology. HbSC patients have a higher hematocrit and greater whole blood viscosity, which produces a state of chronic, sluggish blood flow and indolent, low-grade ischemia. This condition provides a sustained stimulus for the release of proangiogenic factors like vascular endothelial growth factor (VEGF).7 In contrast, the more aggressive sickling in HbSS disease leads to more complete arteriolar occlusions, resulting in profound infarction of the peripheral retina—an environment less conducive to a sustained angiogenic response. This paradox is the central organizing principle for risk stratification in SCR.
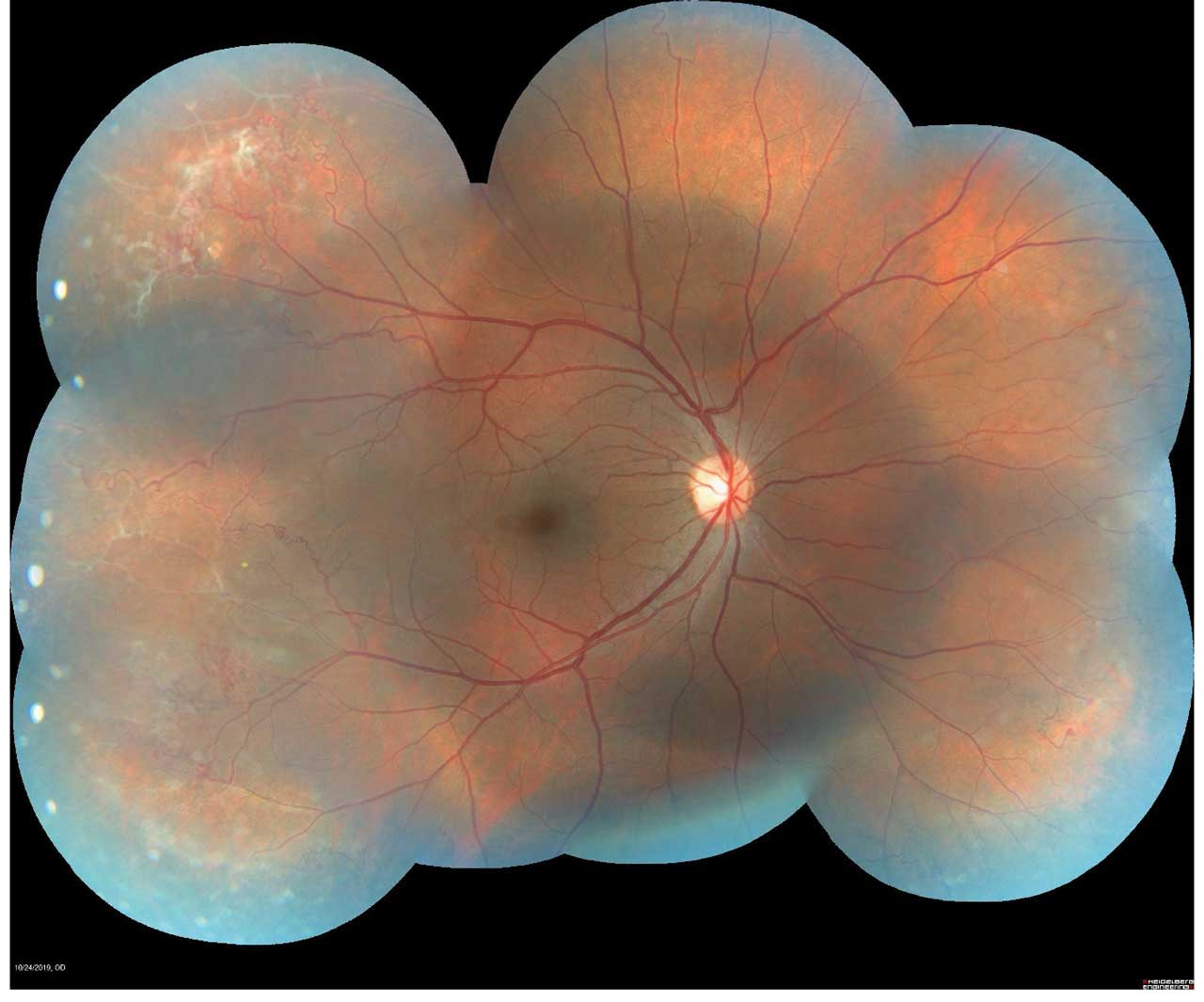
Figure 1. Montage color fundus photograph of proliferative sickle cell retinopathy showing peripheral sea fan neovascularization. Photo by Katie Lachut-Yevich/VCU Health System.
Screening
Evidence-based guidelines advise a baseline dilated fundus examination at age 10, with repeat screening every 1 to 2 years in asymptomatic patients; patients with any retinopathy should be referred to a retina specialist for management and tailored follow-up.8 In practice, risk-stratified intervals are reasonable: HbSC and S-thalassemia (higher PSCR risk) often warrant annual exams, whereas HbSS may be followed every 1-2 years if normal at baseline and low risk.9
Screening remains exam first, but multimodal imaging now improves detection and monitoring (Figure 1).10 SD-OCT helps identify characteristic macular thinning or ischemic changes that may be clinically subtle.11 Ultrawidefield (UWF) color imaging expands peripheral assessment, and OCTA—ideally widefield when available—noninvasively quantifies macular/peripheral perfusion and correlates with disease stage, supporting earlier detection and objective follow-up (Figure 2).12 Fluorescein angiography is reserved for cases where treatment planning (eg, laser) requires defining peripheral nonperfusion or neovascularization.
Classification and Natural History
The clinical spectrum of SCR is divided into nonproliferative (NPSCR) and proliferative stages. NPSCR encompasses retinal findings that result from vaso-occlusion but precede neovascularization. These signs serve as crucial indicators of underlying ischemic activity and include peripheral findings such as salmon-patch hemorrhages, iridescent spots (refractile bodies), and black sunbursts (chorioretinal scars).13
PSCR represents progression to ischemia-driven neovascularization and is most commonly staged using the Goldberg classification system, which provides a framework for clinical staging and management decisions.14,15 Goldberg’s 5-stage system describes disease progression from retinal ischemia to vision-threatening complications:
- Stage I: Peripheral Arteriolar Occlusion. The initial ischemic event that creates areas of peripheral capillary nonperfusion.
- Stage II: Peripheral Arteriovenous Anastomoses. Dilation of preexisting capillary channels at the border between the perfused and nonperfused retina.
- Stage III: “Sea-Fan” Neovascularization. The hallmark of PSCR, defined by the ischemia-driven proliferation of new blood vessel complexes (Figure 3).
- Stage IV: Vitreous Hemorrhage (VH). Bleeding from the fragile sea-fan complexes into the vitreous cavity.
- Stage V: Retinal Detachment (RD). Progressive fibrovascular proliferation leading to retinal detachment.
Recent population-based data have further clarified SCR’s clinical trajectory. In a national electronic health record comparing SCD patients with matched controls without SCD or sickle cell trait, patients with HbSS disease carry a 2.33-fold increased risk of retinal vascular occlusions, including a 2.71-fold higher risk of central retinal artery occlusion and 4.90-fold higher risk of branch retinal artery occlusion.16 These findings suggest that large-vessel arterial events may accompany or even precede Stage I changes, particularly in HbSS genotypes.
Additional insight comes from a registry-based study of 1,249 adults with SCR, which demonstrated a cumulative rise in vision-threatening complications over time.17 Within 5 years of diagnosis, 19.5% of patients developed VH, 12.4% experienced rhegmatogenous retinal detachment (RRD), and 4.7% had tractional retinal detachment (TRD). One notable feature of PSCR is the phenomenon of spontaneous regression, also known as autoinfarction, of sea-fan complexes. This process is estimated to occur in up to 60% of cases and introduces uncertainty regarding the optimal timing of intervention.18
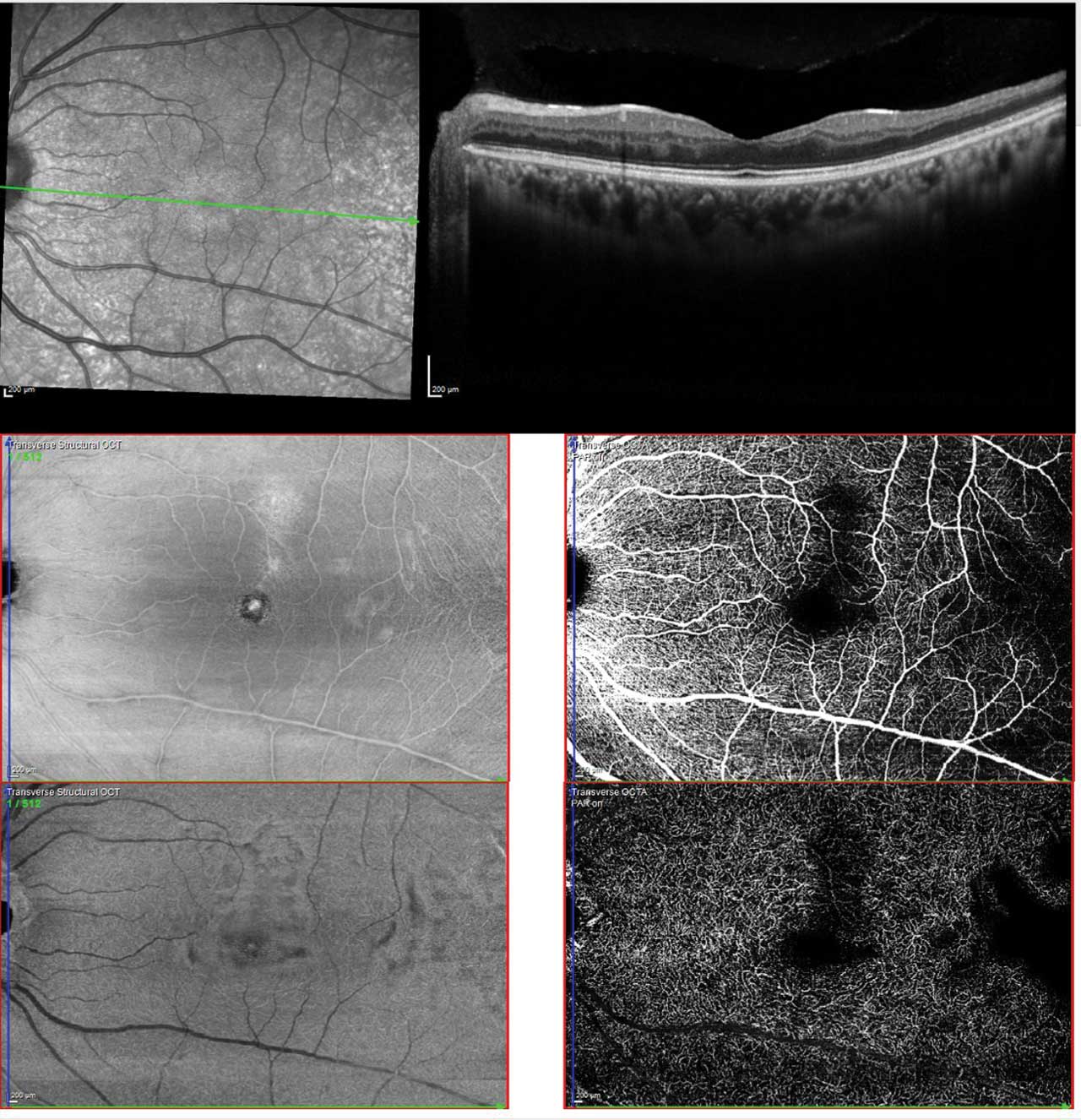
Figure 2. Multimodal imaging of sickle cell maculopathy showing temporal parafoveal thinning on OCT (upper), reduced capillary density in the superficial plexus (middle), and marked capillary dropout in the deep plexus (lower). Photo by Katie Lachut-Yevich/VCU Health System.
Current Standard of Care
Management of SCR is stage-dependent, with observation serving as the cornerstone for both NPSCR and early PSCR disease. The natural history of the disease supports this conservative strategy, because longitudinal cohort studies demonstrate spontaneous regression of neovascularization in up to 32% of eyes, with permanent vision loss being uncommon in early stages.6 Comparative studies for specific subtypes of sea-fan neovascularization have shown no significant difference in visual outcomes between laser-treated and untreated eyes,19 although 13% of untreated eyes developed complications such as VH and retinal detachment.
Scatter laser photocoagulation remains the most performed intervention for high-risk PSCR. In a recent population-based registry study, 13.6% of patients with SCR underwent laser photocoagulation within 5 years of diagnosis.17 Its primary goal is to ablate the ischemic peripheral retina, thereby suppressing the angiogenic stimulus responsible for proliferative disease. In a prospective study of 174 eyes, sectoral scatter photocoagulation significantly reduced the incidence of both VH and vision loss.20 This therapy has largely replaced earlier treatments with higher morbidity, such as feeder-vessel coagulation and cryotherapy. Potential complications include iatrogenic retinal breaks, anterior- segment ischemia, and acute choroidal ischemia.
Pars plana vitrectomy (PPV) is typically reserved for advanced complications such as nonclearing VH, TRD, and RRD. Population-based data reported by Nangia et al show that 6.4% of patients with SCR underwent PPV within 5 years of diagnosis.17 Early case series from the 1970s and 1980s reported modest outcomes, with only 22% to 50% of eyes with TRD/RRD achieving 20/40 or better visual acuity and single operation success rates of 56% to 64%.21-23 The visual outcomes for these complex detachments approximate those reported for diabetic traction retinal detachment, where studies found that vitrectomy improved vision in roughly 59%-70% of eyes but could worsen vision in 15%-34%.24 Modern techniques have markedly improved results: a 2018 study comparing 23-gauge and 20-gauge vitrectomy in 71 eyes with PSCR complications reported mean best corrected visual acuity improving from logMAR 1.30 (≈20/400) to 0.74 (≈20/110), a primary retinal reattachment rate of 79%, and slightly greater visual gain with 23-gauge instrumentation.25 This trend is reinforced by the largest recent case series of 65 eyes, which showed a mean visual acuity improvement from 20/944 to 20/56 in eyes with VH and a single-operation success rate of up to 72% for retinal detachment.26 These improvements are attributed to advances in surgical instrumentation, including small-gauge vitrectomy systems, wide-angle viewing, and valved cannulas, as well as refined techniques like segmentation of fibrovascular membranes and selective use of scleral buckling.26 Despite these gains, iatrogenic retinal breaks remain a concern during membrane delamination.
Scleral buckle (SB) is used infrequently in PSCR, generally limited to select RRD without significant VH or tractional components. Large registry analysis further supports this, showing that only 1.3% of patients with SCR underwent SB within 5 years of diagnosis.17 Historically, SB was considered high-risk for anterior segment ischemia (ASI) due to reduced anterior ciliary artery flow.27 Contemporary series have not shown increased ASI with encirclement, likely reflecting lower-tension, narrower encirclements rather than high, broad bands.24 Because encircling elements can still reduce ocular perfusion, SB is best reserved for younger patients with clear media and discrete peripheral breaks, whereas concurrent fibrovascular proliferation or tractional components more often necessitate vitrectomy.
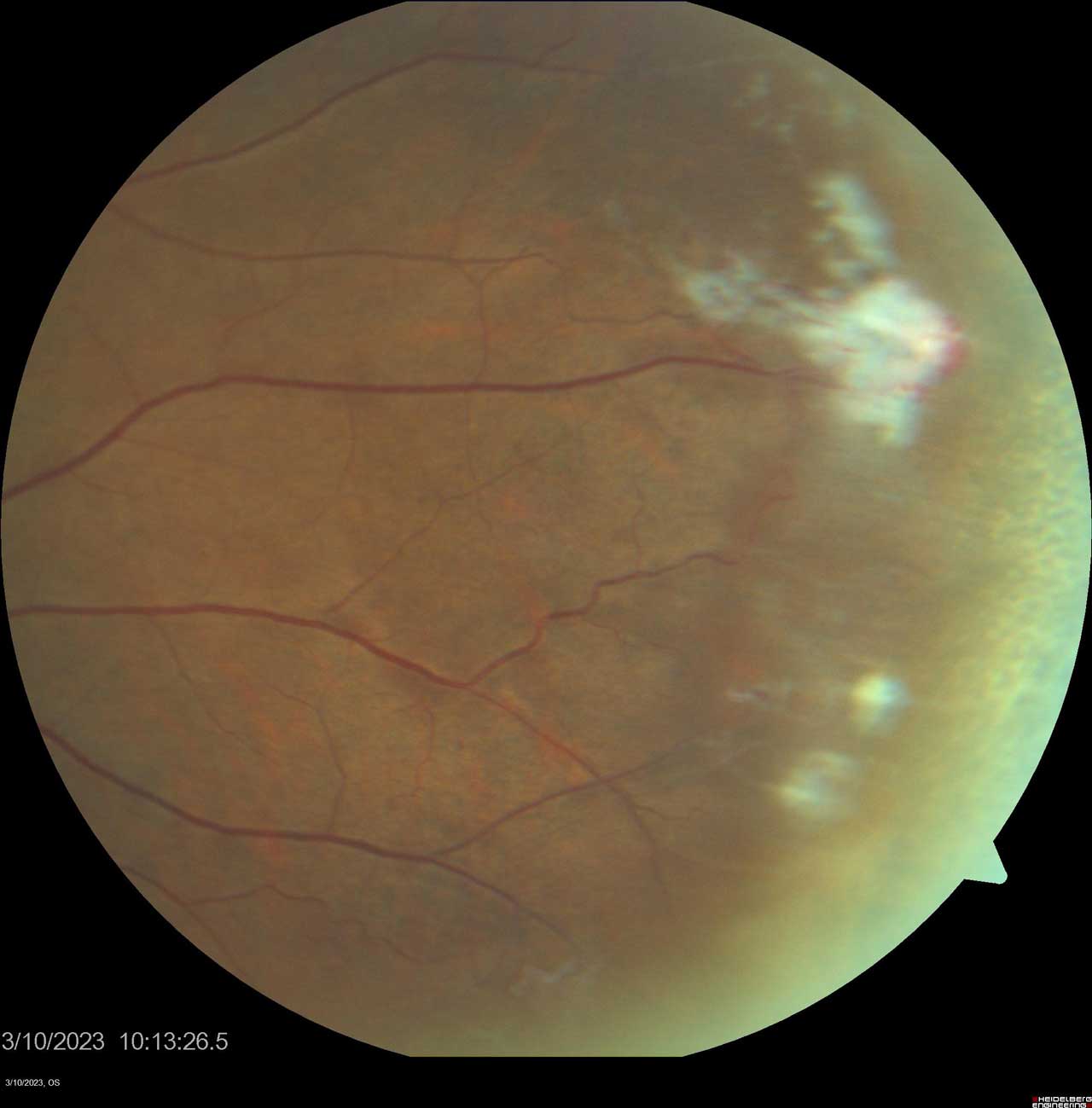
Figure 3. Sea fan neovascularization is a hallmark finding of Stage III proliferative sickle cell retinopathy in the Goldberg classification system. Photo by Katie Lachut-Yevich/VCU Health System.
Intravitreal Anti-VEGF Therapy
Anti-VEGF agents, although off-label for PSCR, have increasingly been employed as adjuncts in Stage III and Stage IV disease. The evidence was initially limited to small retrospective series, which showed that intravitreal bevacizumab could induce regression of sea-fan neovascularization and facilitate clearance of VH.28-31 For example, one early series of 5 patients reported that bevacizumab led to partial neovascular regression and visual acuity improvement in patients with VH.28 More recently, the largest series to date, a multicenter study of 45 eyes, has provided stronger evidence for this approach.32 The study found that in eyes with Stage IV disease, median visual acuity improved markedly from 20/200 to 20/30, and that anti-VEGF therapy controlled the disease without vitrectomy in 93% of all cases.
Clinically, anti-VEGF is most useful when neovascularization persists after laser, when hemorrhage precludes delivery of photocoagulation, or as a preoperative measure before vitrectomy. Administering an anti-VEGF injection approximately 3 to 7 days before surgery may cause significant vascular regression of sea fans, making fibrovascular membranes less friable and minimizing the risk of intraoperative hemorrhage.29 The transient nature of its effect limits its utility as monotherapy, and although theoretical concerns for a tractional “crunch” retinal detachment exist,33 this is not well documented in PSCR. Notably, the largest series to date supported this safety profile, reporting no cases of retinal tear or tractional retinal detachment among 45 treated eyes.32 At present, anti-VEGF therapy is not considered a primary monotherapy but serves as a valuable adjunctive tool in select cases.
Systemic Therapies and Retinal Impact
Hydroxyurea remains the most extensively studied treatment for SCD, reliably increasing fetal hemoglobin (HbF) to dilute HbS, reduce adhesion molecule expression, and dampen vaso-occlusion. Retrospective pediatric and adult cohorts have demonstrated that HbF levels above ~15% are associated with a 50% reduction in the risk of developing SCR.34,35 In one pediatric study, children with HbF below 15% were more than 7 times more likely to develop SCR than those with HbF near 16%.34 Hydroxyurea has also been associated with reduced retinal thinning observed on OCT and improved perfusion metrics, including decreased intermittent perfusion index on OCTA.11 However, no randomized controlled trials have prospectively included retinal endpoints.
Several other systemic agents have shown benefits in reducing vaso-occlusive events, but their impact on the retina remains unknown. Voxelotor, a hemoglobin polymerization inhibitor, increased hemoglobin by approximately 1 g/dL and reduced hemolysis in its pivotal program (HOPE),36,37 but it was voluntarily withdrawn globally in September 2024; no ophthalmic endpoints were assessed, so any retinal benefit remains unproven. Crizanlizumab, an anti-P-selectin inhibitor, reduced vaso-occlusive crises in the SUSTAIN trial and received US Food and Drug Administration (FDA) approval in 2019.38 However, the STAND confirmatory trial did not reproduce this efficacy signal, leading to the withdrawal of its EU and UK authorizations; again, no visual or imaging endpoints were included. L-glutamine (Endari; Emmaus Medical) has FDA approval from 2017, based on a randomized trial showing fewer pain crises and hospitalizations,39 but no retinal outcomes were captured.
Gene Therapy and Curative Approaches
The recent FDA approvals of exagamglogene autotemcel (Casgevy; Vertex Pharmaceuticals) and lovotibeglogene autotemcel (Lyfgenia; Bluebird Bio) mark a new era in the treatment of SCD.40 Casgevy reactivates fetal hemoglobin via CRISPR/Cas9 editing of the BCL11A erythroid enhancer, while Lyfgenia uses a lentiviral vector to insert an anti-sickling β-globin transgene. Both therapies aim to eliminate sickling at its source, and early data are encouraging: 97% of patients in the CLIMB-121 trial receiving Casgevy remained free of vaso-occlusive events for at least 12 months,41 while 88% of patients receiving Lyfgenia in a separate trial achieved similar outcomes.42 However, neither study incorporated ophthalmic imaging or vision-specific endpoints, and the impact of these therapies on SCR remains unknown.
Allogeneic hematopoietic stem cell transplantation (HSCT) remains the only established cure for SCD, halting systemic progression in most patients.43 However, its effect on ocular outcomes is more limited. In a 2025 prospective study, SCR progression occurred in only 1.7% of HSCT patients vs 15% of controls (P=.026), yet neovascularization still developed in 1 case despite full donor chimerism.44 Macular thinning also progressed in 7.1% of HSCT eyes, likely reflecting preexisting microvascular damage. These findings suggest HSCT may reduce, but not fully prevent, ocular complications.
Future Directions
The management of SCR is entering a new era, shaped by curative systemic therapies and rapid advancements in diagnostic imaging. Moving forward, well-designed randomized trials are needed to compare imaging-guided laser, anti-VEGF therapy, and structured observation protocols using modern endpoints. Advanced imaging offers a noninvasive, reproducible platform for tracking peripheral ischemia and may serve as a trial-ready biomarker of disease activity.10,12 Meanwhile, AI and deep learning tools show promise in automating detection, predicting progression risk, and expanding access to care.45 Together, these innovations set the stage for a proactive, personalized, and evidence-based approach to preserving vision in patients with SCD. RP



References
1. National Heart, Lung, and Blood Institute. What is sickle cell disease? National Institutes of Health. September 30, 2024. Accessed September 5, 2025. https://www.nhlbi.nih.gov/health/sickle-cell-disease
2. GBD 2021 Sickle Cell Disease Collaborators. Global, regional, and national prevalence and mortality burden of sickle cell disease, 2000-2021: a systematic analysis from the Global Burden of Disease Study 2021. Lancet Haematol. 2023;10(8):e585-e599. doi:10.1016/S2352-3026(23)00118-7
3. US Centers for Disease Control. Data and statistics on sickle cell disease. May 22, 2024. Accessed September 5, 2025. https://www.cdc.gov/sickle-cell/data/index.html
4. Anibire OO, Brill DA, Jr BKW. Sickle cell retinopathy and systemic disease. Annals of Eye Science. 2024;9(0):11-11. doi:10.21037/aes-23-8
5. American Academy of Ophthalmology. Management of proliferative sickle cell retinopathy. September 25, 2018. Accessed September 17, 2025. https://www.aao.org/eyenet/article/proliferative-sickle-cell-retinopathy
6. Downes SM, Hambleton IR, Chuang EL, Lois N, Serjeant GR, Bird AC. Incidence and natural history of proliferative sickle cell retinopathy: observations from a cohort study. Ophthalmology. 2005;112(11):1869-1875. doi:10.1016/j.ophtha.2005.05.026
7. Fadugbagbe AO, Gurgel RQ, Mendonça CQ, Cipolotti R, dos Santos AM, Cuevas LE. Ocular manifestations of sickle cell disease. Ann Trop Paediatr. 2010;30(1):19-26. doi:10.1179/146532810X12637745451870
8. Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA. 2014;312(10):1033-1048. doi:10.1001/jama.2014.10517
9. Smith BD, Hankins JS, Kang G, et al. Investigation of sickle cell retinopathy in pediatric and adolescent patients enrolled in a large cohort study. Ophthalmology. 2025;132(8):911-920. doi:10.1016/j.ophtha.2025.03.031
10. Hoyek S, Chaaya C, Lemire CA, et al. Retinal imaging biomarkers and correlation to systemic disease activity in pediatric sickle cell disease. Ophthalmol Sci. 2025;5(5):100774. doi:10.1016/j.xops.2025.100774
11. Lim JI, Niec M, Sun J, Cao D. Longitudinal assessment of retinal thinning in adults with and without sickle cell retinopathy using spectral-domain optical coherence tomography. JAMA Ophthalmology. 2021;139(3):330-337. doi:10.1001/jamaophthalmol.2020.6525
12. Clarke K, Mannath A, Anastasi M, et al. Optical coherence tomography angiography as a tool for diagnosis and monitoring of sickle cell related eye disease: a systematic review and meta-analysis. Eye. 2025;39(11):2112-2123. doi:10.1038/s41433-025-03814-1
13. Elagouz M, Jyothi S, Gupta B, Sivaprasad S. Sickle cell disease and the eye: old and new concepts. Surv Ophthalmol. 2010;55(4):359-377. doi:10.1016/j.survophthal.2009.11.004
14. Goldberg MF. Natural history of untreated proliferative sickle retinopathy. Arch Ophthalmol. 1971;85(4):428-437. doi:10.1001/archopht.1971.00990050430006
15. Goldberg MF. Retinal neovascularization in sickle cell retinopathy. Trans Sect Ophthalmol Am Acad Ophthalmol Otolaryngol. 1977;83(3 Pt 1):OP409-431.
16. Kaufmann GT, Russell M, Shukla P, Singh RP, Talcott KE. Retrospective cohort study of sickle cell disease and large vessel retinal vascular occlusion risk in a national United States database. Ophthalmology Retina. 2025;9(1):46-51. doi:10.1016/j.oret.2024.07.013
17. Nangia P, Wai KM, Scott AW, Rahimy E, Mruthyunjaya P. Ophthalmic outcomes and management of retinopathy in patients with sickle cell disease: a comprehensive health registry study and review of management strategies. Ophthalmic Surg Lasers Imaging Retina. 2025;56(8):488-493. doi:10.3928/23258160-20250425-01
18. Lutty GA, McLeod DS, Goldberg MF. Sickle cell retinopathy and hemoglobinopathies. In: Retinal Vascular Diseases. Essentials in Ophthalmology. Springer-Verlag; 2007:273-295. doi:10.1007/978-3-540-29542-6_27
19. Sayag D, Binaghi M, Souied EH, et al. Retinal photocoagulation for proliferative sickle cell retinopathy: a prospective clinical trial with new sea fan classification. Eur J Ophthalmol. 2008;18(2):248-254. doi:10.1177/112067210801800213
20. Farber MD, Jampol LM, Fox P, et al. A randomized clinical trial of scatter photocoagulation of proliferative sickle cell retinopathy. Arch Ophthalmol. 1991;109(3):363-367. doi:10.1001/archopht.1991.01080030065040
21. Pulido JS, Flynn HW, Clarkson JG, Blankenship GW. Pars plana vitrectomy in the management of complications of proliferative sickle retinopathy. Arch Ophthalmol. 1988;106(11):1553-1557. doi:10.1001/archopht.1988.01060140721042
22. Freilich DB, Seelenfreund MH. Hyperbaric oxygen, retinal detachment, and sickle cell anemia. Arch Ophthalmol. 1973;90(2):90-93. doi:10.1001/archopht.1973.01000050092002
23. Jampol LM, Green JL, Goldberg MF, Peyman GA. An update on vitrectomy surgery and retinal detachment repair in sickle cell disease. Arch Ophthalmol. 1982;100(4):591-593. doi:10.1001/archopht.1982.01030030593008
24. Chen RWS, Flynn HW, Lee WH, et al. Vitreoretinal management and surgical outcomes in proliferative sickle retinopathy: a case series. Am J Ophthalmol. 2014;157(4):870-875.e1. doi:10.1016/j.ajo.2013.12.019
25. Ho J, Grabowska A, Ugarte M, Muqit MM. A comparison of 23-gauge and 20-gauge vitrectomy for proliferative sickle cell retinopathy—clinical outcomes and surgical management. Eye (Lond). 2018;32(9):1449-1454. doi:10.1038/s41433-018-0127-y
26. Rohowetz LJ, Panneerselvam S, Williams BK, et al. Proliferative sickle cell retinopathy: outcomes of vitreoretinal surgery. Ophthalmol Retina. 2024;8(8):832-837. doi:10.1016/j.oret.2024.01.023
27. Ryan SJ, Goldberg MF. Anterior segment ischemia following scleral buckling in sickle cell hemoglobinopathy. Am J Ophthalmol. 1971;72(1):35-50. doi:10.1016/0002-9394(71)91588-1
28. Cai CX, Linz MO, Scott AW. Intravitreal bevacizumab for proliferative sickle retinopathy: a case series. J Vitreoretinal Dis. 2018;2(1):32-38. doi:10.1177/2474126417738627
29. Moshiri A, Ha NK, Ko FS, Scott AW. Bevacizumab presurgical treatment for proliferative sickle-cell retinopathy-related retinal detachment. Retin Cases Brief Rep. 2013;7(3):204-205. doi:10.1097/ICB.0b013e3182845d31
30. Siqueira RC, Costa RA, Scott IU, Cintra LP, Jorge R. Intravitreal bevacizumab (Avastin) injection associated with regression of retinal neovascularization caused by sickle cell retinopathy. Acta Ophthalmol Scand. 2006;84(6):834-835. doi:10.1111/j.1600-0420.2006.00779.x
31. Shaikh S. Intravitreal bevacizumab (Avastin) for the treatment of proliferative sickle retinopathy. Indian J Ophthalmol. 2008;56(3):259. doi:10.4103/0301-4738.40380
32. Lim JI, Okonkwo ON, Ong SS, et al. Anti–vascular endothelial growth factor therapy for stages 3 and 4 proliferative sickle cell retinopathy results in improved anatomic and visual outcomes. Retina. Published online August 27, 2025. doi:10.1097/IAE.0000000000004658
33. Cai CX, Scott AW. Surgical approach to sickle cell retinopathy. Retina Today. April 2017. Accessed September 17, 2025. https://retinatoday.com/articles/2017-apr/retina-pearls-surgical-approach-to-sickle-cell-retinopathy
34. Estepp JH, Smeltzer MP, Wang WC, Hoehn ME, Hankins JS, Aygun B. Protection from sickle cell retinopathy is associated with elevated HbF levels and hydroxycarbamide use in children. Br J Haematol. 2013;161(3):402-405. doi:10.1111/bjh.12238
35. Mian UK, Tang J, Allende APM, et al. Elevated fetal haemoglobin levels are associated with decreased incidence of retinopathy in adults with sickle cell disease. Br J Haematol. 2018;183(5):807-811. doi:10.1111/bjh.15617
36. Howard J, Hemmaway CJ, Telfer P, et al. A phase 1/2 ascending dose study and open-label extension study of voxelotor in patients with sickle cell disease. Blood. 2019;133(17):1865-1875. doi:10.1182/blood-2018-08-868893
37. Vichinsky E, Hoppe CC, Ataga KI, et al. A phase 3 randomized trial of voxelotor in sickle cell disease. N Engl J Med. 2019;381(6):509-519. doi:10.1056/NEJMoa1903212
38. Ataga KI, Kutlar A, Kanter J, et al. Crizanlizumab for the prevention of pain crises in sickle cell disease. N Engl J Med. 2017;376(5):429-439. doi:10.1056/NEJMoa1611770
39. Niihara Y, Miller ST, Kanter J, et al. A phase 3 trial of l-glutamine in sickle cell disease. N Engl J Med. 2018;379(3):226-235. doi:10.1056/NEJMoa1715971
40. US Food and Drug Administration. FDA approves first gene therapies to treat patients with sickle cell disease. August 9, 2024. Accessed September 17, 2025. https://www.fda.gov/news-events/press-announcements/fda-approves-first-gene-therapies-treat-patients-sickle-cell-disease
41. Frangoul H, Locatelli F, Sharma A, et al. Exagamglogene autotemcel for severe sickle cell disease. N Engl J Med. 2024;390(18):1649-1662. doi:10.1056/NEJMoa2309676
42. Kanter J, Walters MC, Krishnamurti L, et al. Biologic and clinical efficacy of LentiGlobin for sickle cell disease. N Engl J Med. 2022;386(7):617-628. doi:10.1056/NEJMoa2117175
43. Kanter J, Liem RI, Bernaudin F, et al. American Society of Hematology 2021 guidelines for sickle cell disease: stem cell transplantation. Blood Adv. 2021;5(18):3668-3689. doi:10.1182/bloodadvances.2021004394C
44. Brandsen RP, Dovern E, Biemond BJ, Diederen RMH, Nur E. The impact of allogeneic hematopoietic stem cell transplantation on sickle cell retinopathy and maculopathy: a prospective, observational study. Am J Hematol. 2025;100(8):1448-1452. doi:10.1002/ajh.27705
45. Cai S, Han IC, Scott AW. Artificial intelligence for improving sickle cell retinopathy diagnosis and management. Eye (Lond). 2021;35(10):2675-2684. doi:10.1038/s41433-021-01556-4