







As retina specialists, we stand at the forefront of some of the most innovative therapeutic developments in medicine. From anti-VEGF agents that revolutionized neovascular age-related macular degeneration (nAMD) treatment to emerging gene therapies for inherited retinal diseases, our specialty has benefited tremendously from rigorous clinical research. Having served as both a clinical investigator and in executive roles at biotech companies, I have witnessed firsthand how the quality of site-sponsor collaboration can make or break a clinical trial’s success.
The foundation of successful collaboration rests on the commitments we make when signing US Food and Drug Administration (FDA) Form 1572, our Statement of Investigator. These aren’t merely bureaucratic requirements—they represent the ethical and operational framework that ensures patient safety, data integrity, and ultimately, the advancement of retinal medicine. By signing Form 1572, investigators agree to 9 specific obligations that form the foundation of ethical clinical research:
- Protocol Compliance: Conduct studies according to the current protocol and notify sponsors of any changes, except when necessary to protect subject safety, rights, or welfare;
- Personal Supervision: Personally conduct or supervise all described investigations;
- Informed Consent: Inform patients that drugs are investigational and ensure compliance with informed consent requirements (21 CFR Part 50) and institutional review board (IRB) review/approval (21 CFR Part 56);
- Adverse Event Reporting: Report adverse experiences to sponsors in accordance with 21 CFR 312.64;
- Investigator Brochure Review: Read and understand all information in the investigator’s brochure, including potential risks and side effects;
- Team Education: Ensure all associates, colleagues, and employees understand their obligations in meeting these commitments;
- Record Maintenance: Maintain adequate and accurate records per 21 CFR 312.62 and make them available for inspection per 21 CFR 312.68;
- IRB Oversight: Ensure compliant IRB responsibility for initial and continuing review, promptly report changes and unanticipated problems, and obtain IRB approval before making research changes except to eliminate immediate hazards; and
- Regulatory Compliance: Comply with all other clinical investigator obligations and pertinent requirements in 21 CFR Part 312.
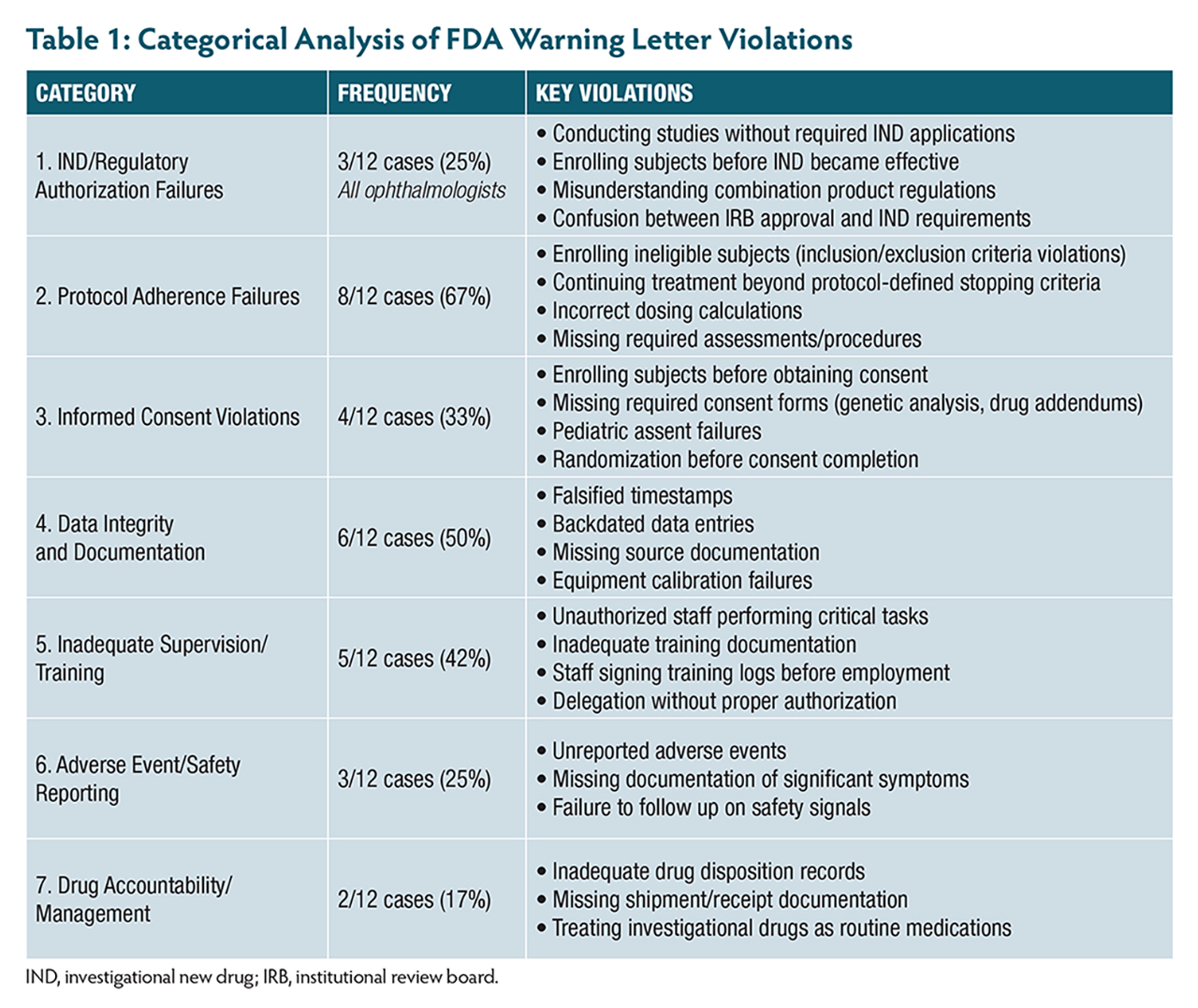
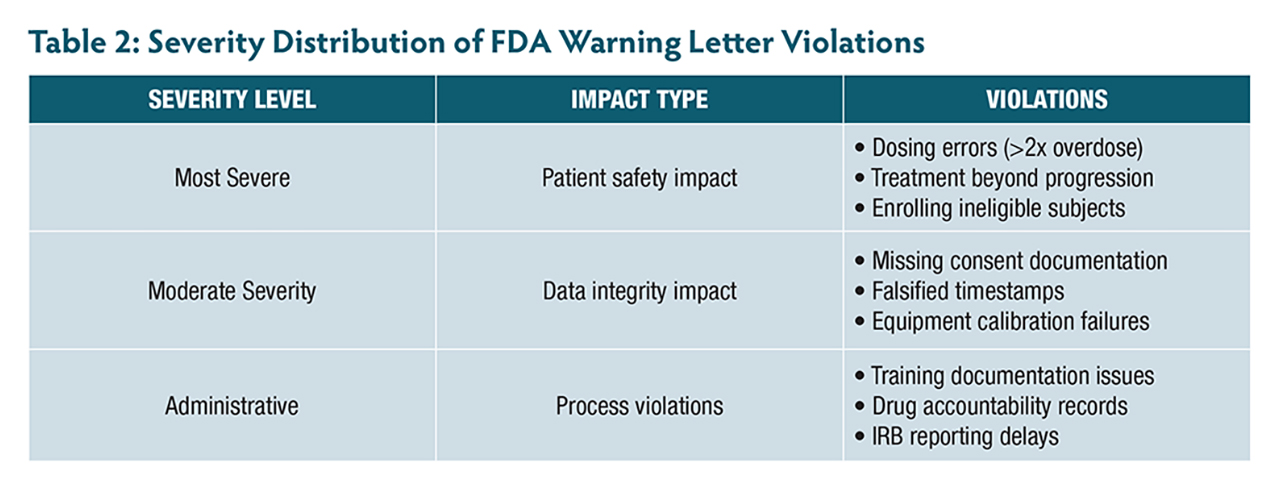
A review of 12 FDA warning letters issued between 2021 and 2025—including 3 to ophthalmologists—highlights the real-world consequences of not adhering to these principles. These cases reveal patterns of violations (Table 1, Table 2) that are both preventable and particularly relevant to retinal research.
Protocol Adherence and Investigator Supervision
The first commitment in Form 1572 states: “I agree to conduct the study(ies) in accordance with the relevant, current protocol(s)” and “I agree to personally conduct or supervise the described investigation(s).” Recent FDA enforcement actions reveal how easily investigators stumble on these fundamental requirements.
One investigator enrolled subjects with active infections despite clear exclusion criteria, while an ophthalmologist continued treating patients with investigational therapy despite confirmed disease progression that met protocol-defined criteria for treatment discontinuation. Even more alarming, 1 site administered more than double the intended investigational product dose because staff incorrectly documented weight as 152 kg instead of 152 lb (68.9 kg). The investigator had signed the prescription electronically but failed to verify the calculation—a potentially catastrophic oversight in research, where precise dosing is paramount.
Another case involved unauthorized staff performing critical study tasks without proper authorization, with training records showing staff had signed protocol training logs dated 3 months before their actual employment started.
In retinal research, this balance becomes particularly nuanced. Consider a patient enrolled in a geographic atrophy (GA) study who develops new onset nAMD in the study eye. Although immediate anti-VEGF treatment may be necessary, the key is immediate communication with the sponsor and documentation of your rationale.
To maintain protocol adherence and ensure effective investigator supervision, the following strategies are critical:
- Document safety-related deviations within 24 hours with clear clinical rationale;
- Personally review all dosing calculations and prescriptions before administration;
- Maintain current delegation logs with clearly defined roles and verify training completion; and
- Be especially vigilant with progression criteria in retinal studies where anatomic changes may be subtle.
Informed Consent and Special Populations
The commitment to ensure informed consent compliance under 21 CFR Part 50 goes far beyond obtaining a signature. Recent FDA actions reveal common pitfalls, including subjects randomized before consent was obtained and an ophthalmologist enrolling 6 minors (ages 11 to 17) without obtaining required written assent, despite IRB requirements for documented assent from children in this age range.
Another investigator failed to obtain required separate consent forms for genetic analysis, collecting blood samples at multiple timepoints without proper authorization. When discovered, the investigator had to request destruction of the samples and notify the IRB.
In retinal research, we often deal with patients facing vision loss—a profoundly emotional situation affecting comprehension and decision-making. For inherited retinal diseases affecting children and families, the consent process becomes even more complex. Consider a patient with Stargardt disease considering gene therapy—they need to understand not just the injection risks, but the long-term uncertainty about efficacy and available alternatives.
To ensure ethical and effective participation, the following strategies focus on informed consent and accommodating the unique considerations of special populations:
- Use visual aids and diagrams to explain complex retinal procedures;
- Ensure all required consent forms are obtained, including separate forms for genetic analysis;
- For pediatric patients, obtain proper written assent in addition to parental consent; and
- Document comprehension verification, not just signature completion.
IND Requirements: A Critical Knowledge Gap in Ophthalmology
One of the most alarming patterns involves 3 ophthalmologists conducting clinical investigations without proper investigational new drug (IND) applications. One corneal specialist conducted a crosslinking study using riboflavin solutions combined with UVA light without submitting an IND, enrolling 32 subjects in what the investigator believed was an IRB-approved device study. Another enrolled and treated 4 subjects before the IND was in effect, mistakenly believing IRB approval meant the IND was also approved.
A third case involved an ophthalmologist who believed a combination product study qualified as a nonsignificant risk device study, when the drug components required IND oversight. Even after IRB notification that an IND was required, the investigator submitted an IND for a different protocol than the one being conducted.
For retinal specialists, this issue is particularly relevant for combination product studies, investigator-initiated trials with off-label medications, novel drug delivery methods, and gene therapy studies involving vector delivery systems.
Investigators should keep these critical IND compliance requirements in mind when planning and conducting retinal studies:
- Consult with FDA or regulatory experts before initiating any investigator-sponsored study;
- Understand that IRB approval does not substitute for IND requirements;
- Remember that INDs become effective 30 days after FDA receipt, not upon submission; and
- Ensure proper coverage for combination products commonly used in retinal research.
Adverse Event Reporting and Documentation
Recent enforcement actions reveal investigators failing to report vomiting after procedures, significant postsurgical pain, and various symptoms noted in progress notes but never documented as adverse events (AEs). In retinal research, distinguishing between expected disease progression and true adverse events requires clinical expertise. The FDA emphasizes using recognized medical terminology and documenting specific visual acuity measurements, visual field changes, and clinical findings.
Another serious pattern involves data integrity violations. One investigator’s site had approximately 150 measurements potentially compromised by an uncalibrated instrument over 12 months, affecting 32 subjects. Another case involved falsified timestamps where assessment data was entered days or weeks after visits but backdated to appear contemporaneous.
To uphold the highest standards in retinal research, investigators should follow these key practices for adverse event reporting and documentation:
- Develop standard operating procedures for AE identification in retinal studies;
- Maintain proper calibration and maintenance of all imaging equipment;
- Ensure contemporaneous documentation of all retinal imaging and functional testing; and
- Never backdate entries or alter timestamps in electronic systems.
Excellence in Site-Sponsor Collaboration
Although regulatory compliance forms the foundation, true excellence emerges from ongoing collaboration quality. The most successful retinal research sites understand their relationship with sponsors extends from initial feasibility assessment through final database lock.
Pre-study excellence: Provide realistic feasibility assessments. For a GA study, do not simply count all GA patients—consider specific lesion characteristics, visual acuity requirements, and exclusionary conditions. A realistic assessment might reveal that your initial estimate of 50 potential subjects represents only 15 to 20 truly eligible candidates. Account for seasonal patterns, scheduled absences, and competing studies in your projections.
During-study excellence: Respond to queries within 48 to 72 hours and communicate proactively about potential issues. If your optical coherence tomography (OCT) machine requires unexpected calibration or a key staff member leaves, early communication allows collaborative problem-solving rather than crisis management.
Data lock excellence: When case report forms are ready for final review and signature, treat this as priority. Sponsors may have spent months preparing for database lock, coordinating with statistical teams and regulatory consultants. Delays can cascade through the entire project timeline, potentially delaying regulatory submissions.
Quality over quantity: Sponsors consistently prefer responsive investigators who deliver high-quality data over high-volume recruiters with poor responsiveness. A site enrolling 15 patients with clean data, minimal queries, and prompt communication provides more value than a site enrolling 30 patients requiring extensive data cleaning and creating project management challenges.
Special Considerations for Retinal Research
Retinal research presents unique challenges requiring additional attention to fundamental principles. The complexity of retinal imaging interpretation requires investigators to personally review critical assessments rather than delegating interpretation to coordinators.
Subretinal injections involve complex surgical procedures with specific safety considerations, so investigator understanding of and adherence to protocol becomes even more critical. For inherited retinal diseases, pediatric consent issues require specialized expertise, and precise dosing verification procedures are essential for intraocular injections.
Building Sustainable Programs and Looking Forward
Excellence in clinical research requires systematic approaches sustained over multiple studies and years. The most successful retinal research sites develop comprehensive standard operating procedures, invest in ongoing staff training including imaging technique certification, and create quality metrics for critical measurements.
This systematic approach pays dividends in sponsor relationships. Sites that consistently deliver high-quality data with minimal queries receive preferential consideration for new studies and selection for complex or prestigious research programs. The warning letters we examined show the consequences of failing to maintain these standards—investigators face enforcement action and lose opportunities to participate in future studies, ultimately harming patient access to innovative treatments.
Conclusion: Excellence as a Choice
Every time we sign Form 1572, we make a choice to conduct research meeting the highest standards of scientific rigor and ethical integrity. Our examination of 12 randomly selected FDA warning letters, including 3 directed at ophthalmologists, serves as a sobering reminder of what happens when investigators fail to uphold these standards.
The commitments in Form 1572 are not burdens imposed by regulatory bureaucracy—they are professional standards that define us as clinical investigators. The ophthalmology warning letters reveal that our specialty faces unique challenges requiring specialized knowledge and attention. From IND requirement misunderstandings to critical dosing errors, from inadequate informed consent to data integrity violations, these cases demonstrate that regulatory requirements are not bureaucratic obstacles—they are patient safety imperatives.
As we continue pushing the boundaries of what is possible in retinal medicine, let’s ensure our research practices are worthy of the trust placed in us by patients and the broader medical community. The future of retinal medicine depends not just on innovative science, but also on the excellence with which we conduct clinical research that transforms scientific discoveries into therapeutic realities. Our patients—and the future of retinal medicine—deserve nothing less than our commitment to the highest standards of clinical research conduct. RP
